Mon, Jul 6, 2026
Volume 11 - Continuous Publishing
Iran J Neurosurg 2025, 11 - Continuous Publishing: 0-0 |
Back to browse issues page

Ethics code: IR.GUMS.REC.1403.037
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Abbasi H, Morovat S, Yousefzadeh-Chabok S, Norollahi S E, Nejatifar F, Delpasand K, et al . Bioinformatics-based Study of Cancer Stem Cell Function as Diagnostic Biomarkers in Glioblastoma Multiforme. Iran J Neurosurg 2025; 11 : 22
URL: http://irjns.org/article-1-500-en.html
URL: http://irjns.org/article-1-500-en.html
Haniye Abbasi 

 , Saman Morovat , Shahrokh Yousefzadeh-Chabok1 , Seyedeh Elham Norollahi , Fatemeh Nejatifar , Kourosh Delpasand2 , Kosar Babaei , Afshin Dalili3 , Ali Akbar Samadani *4
, Saman Morovat , Shahrokh Yousefzadeh-Chabok1 , Seyedeh Elham Norollahi , Fatemeh Nejatifar , Kourosh Delpasand2 , Kosar Babaei , Afshin Dalili3 , Ali Akbar Samadani *4
, Saman Morovat , Shahrokh Yousefzadeh-Chabok1 , Seyedeh Elham Norollahi , Fatemeh Nejatifar , Kourosh Delpasand2 , Kosar Babaei , Afshin Dalili3 , Ali Akbar Samadani *4
1- Guilan Road Trauma Research Center, Trauma Institute, Guilan University of Medical Sciences, Rasht, Iran. & Neuroscience Research Center, Trauma Institute, Guilan University of Medical Sciences, Rasht, Iran.
2- Department of Medical Ethics, Guilan University of Medical Sciences, Rasht, Iran.
3- Guilan Road Trauma Research Center, Trauma Institute, Guilan University of Medical Sciences, Rasht, Iran
4- Guilan Road Trauma Research Center, Trauma Institute, Guilan University of Medical Sciences, Rasht, Iran ,a.a.hormoz@gmail.com
2- Department of Medical Ethics, Guilan University of Medical Sciences, Rasht, Iran.
3- Guilan Road Trauma Research Center, Trauma Institute, Guilan University of Medical Sciences, Rasht, Iran
4- Guilan Road Trauma Research Center, Trauma Institute, Guilan University of Medical Sciences, Rasht, Iran ,
Keywords: Glioblastoma multiforme, bioinformatics, cancer stem cells, glioma cancer cells, signaling pathway
Full Text [PDF 3073 kb]
(224 Downloads)
| Abstract (HTML) (863 Views)
Full Text: (213 Views)
1. Introduction
Glioblastoma multiforme (GBM) is the most aggressive and lethal primary brain tumor in adults, accounting for nearly half of all malignant gliomas [1, 2]. Despite current multimodal treatments, including maximal surgical resection, radiotherapy, and temozolomide chemotherapy, median survival remains poor at approximately 12–15 months, and one-year survival rates are under 5–10% [3]. GBM can arise de novo (primary GBM) or evolve from lower-grade astrocytomas (secondary GBM), with primary cases comprising over 90% and usually affecting older individuals (~55–60 years) [4] without clinical or histologic evidence of a less malignant precursor lesion (primary glioblastomas.
Distinct genetic alterations, including IDH mutations, EGFR amplification, TERT promoter mutations, and MGMT promoter methylation, have been implicated in the pathogenesis and prognosis of GBM. In addition, intratumoral heterogeneity, immune evasion, and the restrictive blood–brain barrier further complicate treatment and contribute to the poor clinical outcome [5, 6]. As such, novel diagnostic markers and therapeutic strategies remain urgently needed.
A subpopulation of glioma stem-like cells (GSCs) (also referred to as cancer stem cells [CSCs]) has emerged as a key driver of GBM recurrence, resistance to therapy, and aggressive behavior. These cells display enhanced self-renewal and tumorigenic potential, and are enriched in hypoxic microenvironments characterized by necrosis and pseudopalisading architecture. Hypoxia-inducible factors, particularly HIF1α and HIF2α, promote CSC maintenance by activating downstream stem cell regulators such as SOX2, OCT4, and CD133, thereby supporting GBM progression [7, 8]. Moreover, other molecular processes (like EMT, epigenetic reprogramming, m6A RNA methylation, and expression of lncRNA H19) also contribute to GSC regulation and therapeutic resistance, underscoring the complexity of GBM biology [8–10].
Current evidence suggests that targeting pathways central to GSC function, such as hypoxia signaling, VEGF, TGF-β, CXCR4, and Notch, hold promise for improving therapeutic outcomes in GBM [6, 7]. However, the translation of CSC-targeted therapies remains challenging due to intratumoral heterogeneity and dynamic interactions within the tumor microenvironment.
Given this context, bioinformatics approaches that integrate gene expression profiling from GBM stem‑like cell models and clinical datasets may uncover novel biomarkers and therapeutic targets. In the current study, we analyze the GEO dataset GSE23806 (comprising glioblastoma stem‑like cell lines and conventional glioma lines) as well as TCGA GBM expression data via the GEPIA2 platform. By identifying common dysregulated genes, examining their prognostic significance, and mapping their protein‑interaction networks, we aim to discover novel diagnostic and survival‑related biomarkers associated with GBM stem‑like biology.
2. Methods and Materials/Patients
Data acquisition and preprocessing
The microarray dataset GSE23806 was downloaded from the Gene Expression Omnibus (GEO) [11] database using the GEOquery package [12, 13] in R software, version 4.2.2. This dataset, based on the Affymetrix Human Genome U133 Plus 2.0 Array platform, includes gene expression profiles from 32 conventional glioma cell lines and 23 glioblastoma stem-like cell lines. The glioblastoma stem-like group comprised 12 GS cell lines, 7 subclonal lines derived from two GS lines, 12 primary tumor samples, and 4 monolayer cultures established from tumors analogous to the GS lines.
Initial quality control and normalization of raw expression data were performed to reduce technical variation and ensure sample comparability. Normalization was confirmed through standard quality assessment metrics and visualizations using boxplots and density plots in R.
Differential expression analysis
To identify differentially expressed genes (DEGs) between glioblastoma stem-like cell lines and conventional glioma cell lines, the limma package from Bioconductor was employed. Genes with a |log₂ fold change ≥1| and an adjusted P<0.01 (Benjamini-Hochberg method) were considered significant.
To validate the results from GSE23806, the Gene Expression Profiling Interactive Analysis 2 (GEPIA2) web server was used to compare gene expression between glioblastoma (GBM) tissues and normal brain tissues, utilizing data from the cancer genome atlas (TCGA). Differentially expressed genes with the same threshold criteria (log₂ fold change ≥1, adj. P<0.01) were retrieved. The intersection of dysregulated genes from both datasets was determined to identify consistently differentially expressed genes in glioblastoma.
Survival analysis
The Kaplan–Meier survival analysis was conducted using GEPIA2 to assess the prognostic significance of the intersected dysregulated genes (n=355). Overall survival (OS) analysis was performed by stratifying patients into high- and low-expression groups based on median gene expression levels. Genes with a log-rank P<0.05 and a Hazard ratio (HR)>0.5 were considered significantly associated with survival outcomes. A manual literature review was conducted using PubMed to identify the top survival-related genes and assess their novelty in the context of GBM.
Protein–protein interaction (PPI) network construction
To explore the functional interactions among survival-associated genes, a PPI network was constructed using the GeneMANIA [14] plugin within the Cytoscape software, version 3.9 [15]. GeneMANIA integrates various data types, including physical protein interactions, co-expression, genetic interactions, co-localization, shared pathways, and protein domain similarity. The network was visualized and analyzed to identify gene clusters and patterns of connectivity.
Functional enrichment analysis
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analyses were performed using the clusterProfiler package [16] in R. The groupGO, enrichGO, and enrichKEGG functions were used to identify significantly enriched biological processes (BPs), molecular functions (MFs), cellular components (CCs), and pathways among the survival-related genes. A q<0.01 (adjusted using the Benjamini-Hochberg method) was set as the threshold for statistical significance.
3. Results
Dataset description and preprocessing
To identify DEGs, the microarray dataset GSE23806 was obtained from the GEO database using the GEOquery package in R. This dataset is based on the Affymetrix human genome U133 Plus 2.0 Array and includes expression profiles from 55 samples, comprising 32 conventional glioma cell lines, and 23 glioblastoma stem-like cell lines (12 GS cell lines, 7 subclonal lines derived from two GS lines, 12 primary tumors, and 4 monolayer cultures established from tumors similar to the GS lines). Before downstream analysis, the dataset underwent quality assessment and normalization to ensure comparability and minimize technical variation (Figure 1).
.PNG)
Identification of dysregulated genes in stem-like and tumor samples
Differential gene expression analysis was conducted using the limma package in R, with thresholds set at a log₂ fold change of ≥1 and an adjusted P<0.01. Based on this, 1211 dysregulated genes were identified in glioblastoma stem-like cell lines compared to conventional glioma cell lines.
To validate and extend these findings, the GEPIA2 web server was utilized to analyze gene expression in TCGA GBM samples in comparison to normal brain tissues. This analysis revealed 5217 genes that were dysregulated in tumor samples. The intersection of the two datasets yielded 355 commonly dysregulated genes, indicating a robust set of glioblastoma-associated transcripts (Figure 2).
.PNG)
Out of 1211 genes dysregulated in GSE23806 and 5217 in GEPIA2, 355 genes were commonly dysregulated, indicating strong concordance between stem-like cell line and tumor tissue expression profiles.
Survival analysis and identification of novel prognostic genes
A Kaplan–Meier survival analysis was performed using GEPIA2 to evaluate the association between OS and the 355 commonly dysregulated genes. As a result, 12 genes were found to have a statistically significant impact on patient survival.
A literature search was subsequently performed via PubMed to assess prior investigations of these 12 genes in glioblastoma. Six of these genes were identified as novel, with no prior evidence linking them to GBM Figure 3, Table 1).
.PNG)
.PNG)
These genes exhibit downregulated expression in GBM tissues compared to stem cell tissues, suggesting their potential as novel diagnostic and prognostic biomarkers for glioblastoma.
Construction of PPI network
To explore the molecular relationships among the 12 survival-related genes, a PPI network was constructed using the GeneMANIA plugin within cytoscape. GeneMANIA incorporates multiple functional datasets, including physical interactions, co-expression, pathways, and shared protein domains.
The resulting network comprised 26 nodes and 256 edges, representing extensive functional connectivity among the survival-associated genes (Figure 4).
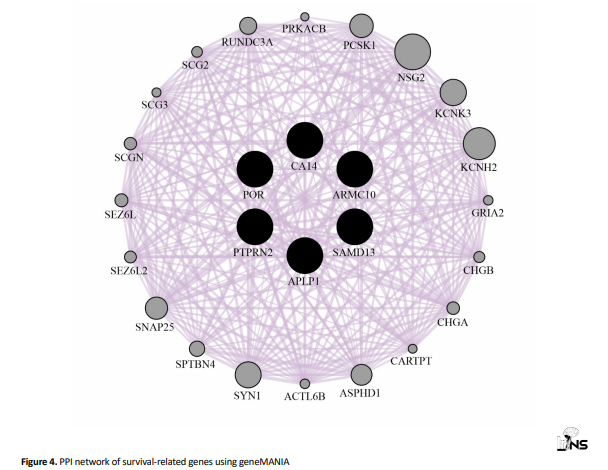
The PPI network reveals a high degree of interconnectivity among the selected genes, indicating potential co-regulatory or synergistic roles in glioblastoma pathogenesis.
Functional enrichment analysis
To gain insight into the biological roles of the survival-related genes, functional enrichment analysis was performed using the clusterProfiler package in R. GO terms and KEGG pathways were assessed using groupGO, enrichGO, and enrichKEGG, with q<0.01 set as the threshold for significance.
This analysis revealed significant enrichment in pathways and BPs related to glioblastoma progression (Figure 5).
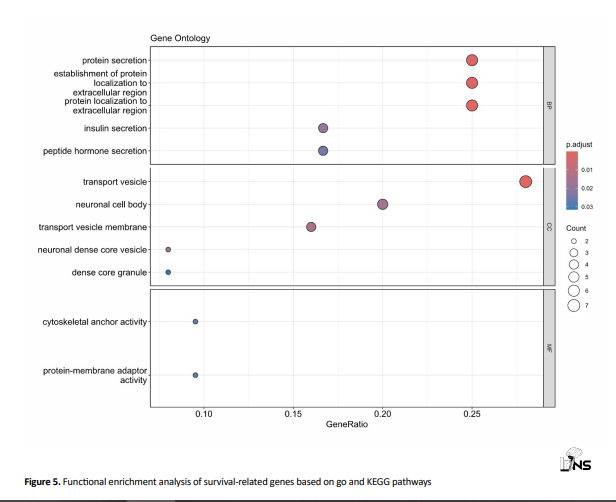
Key enriched GO terms and KEGG pathways highlight the involvement of these genes in critical BPs and signaling pathways relevant to glioblastoma stem cell maintenance and tumorigenesis.
4. Discussion
In this study, we identified 355 genes that are consistently dysregulated in both glioblastoma stem-like cell lines (GSE23806) and TCGA GBM tumor tissues, as determined by GEPIA2, of which 12 genes were significantly associated with overall survival. Remarkably, six genes (APLP1, CA14, PTPRN2, POR, ARMC10, and SMAD13) emerged as novel prognostic markers in GBM, to our knowledge, not previously reported in the context of glioblastoma literature.
In this study, we identified six genes (APLP1, CA14, PTPRN2, POR, ARMC10, and SMAD13) that are dysregulated in glioblastoma compared to stem cells and are significantly associated with patient survival. Based on HRs, two genes (CA14 and SMAD13) appear protective (HR<1), while the other four (APLP1, PTPRN2, POR, and ARMC10) are associated with increased risk (HR>1).
High-risk genes with HR>1 may contribute to more aggressive tumor behavior or stem-like characteristics, driving poorer outcomes. In contrast, protective genes with HR<1 (CA14 and SMAD13) suggest potential tumor-suppressive roles or involvement in pathways that moderate malignancy under hypoxic or stress conditions. This dichotomy reveals the biological complexity of GBM and might highlight context-dependent gene functions, such as differential roles in tumor initiation, progression, or response to therapy.
Although APLP1 has been implicated in neural development and amyloid processing, its diagnostic and prognostic value in GBM is largely unexplored. A recent study found that APLP1-positive extracellular vesicles (EVs) in patient plasma were significantly elevated in GBM, suggesting potential utility as a non-invasive biomarker for early detection [17]. Additionally, network-based analyses have highlighted APLP1, among other genes, as novel candidate biomarkers in GBM pathogenesis. These reports lend credence to our identification of APLP1 as a survival-related gene, supporting further investigation of its role in GBM biology.
CA14 encodes carbonic anhydrase XIV, which plays a crucial role in pH regulatory function, which is essential1818 for maintaining maintaining tumor microenvironment homeostasis under hypoxia and acidosis. Interestingly, CA14 has been identified by machine learning models as a key circadian rhythm gene predictive of melanoma prognosis, providing precedent for its potential as a diagnostic marker across malignancies. Its HR of 0.53 in our GBM cohort suggests that elevated CA14 may contribute to a more regulated microenvironment and improved survival, meriting further investigation in brain tumors [18-20].
POR (cytochrome P450 oxidoreductase) supports metabolic functions essential for the survival of tumor cells. Its association with poorer prognosis (HR=1.7) aligns with the metabolic reprogramming characteristic of aggressive GBM.
ARMC10 knockdown significantly reduced proliferation, migration, invasion, and tumor growth in GBM models. Gene set enrichment associates ARMC10 with Notch signaling and fatty acid metabolism, and the lipid reductions observed upon knockdown underscore its role in metabolic adaptation. Low ARMC10 levels correlated with better patient outcomes, reinforcing its potential as an adverse prognostic marker and therapeutic target.
SMAD13 (like other SMAD family proteins) modulates TGF-β signaling, a pathway heavily implicated in GBM aggression and the mesenchymal subtype. Although less studied than SMAD4 or Smad3, SMAD13 may similarly influence tumor growth, apoptosis, and differentiation. Its HR of 0.56 suggests a possible tumor-suppressive or differentiative role in GBM, potentially antagonizing mesenchymal transition [21-23].
Our PPI network, constructed using GeneMANIA, revealed 26 nodes and 256 edges, underscoring the extensive functional interplay among survival-related genes. Here, the network highlights functional associations, such as pathway co-expression and shared protein domains, suggesting that the novel genes are embedded within broader modules relevant to GBM. Future hub-centric analyses or cluster detection may pinpoint key regulatory modules that are amenable to therapeutic targeting.
The identification of six novel survival‑associated genes provides new avenues for diagnostic and therapeutic research. For instance, APLP1 may be measurable in EVs from blood, representing a minimally invasive diagnostic or monitoring tool. Other genes could serve as therapeutic targets, especially if they mediate stem-like or survival pathways in GBM. However, our findings are based on bioinformatics analyses, and further experimental validation in patient cohorts and functional assays is required. Future studies should evaluate whether these genes are expressed in glioma stem cell populations, respond to hypoxia or treatment, and affect tumor progression in xenograft or spheroid models.
A critical limitation of our work is the absence of experimental validation. Our findings are entirely based on bioinformatics and database-derived analyses and have not yet been corroborated through laboratory assays, such as RT-qPCR, Western blotting, immunohistochemistry, or functional characterization in vitro or in vivo.
This limitation mirrors a common challenge in bioinformatics-based biomarker discovery. For instance, prior studies employing integrated expression and survival analyses often emphasized the need for subsequent laboratory validation to confirm predicted biomarkers [24, 25]. Similarly, translational model investigations have shown that while in silico models may yield promising targets, only through direct experimental validation (such as gene knockdown or protein-level validation) can translational relevance be established [26].
5. Conclusion
By integrating stem-like GBM cell line data with tumor expression profiles and survival analysis, this study uncovered novel biomarker candidates (notably APLP1, CA14, PTPRN2, POR, ARMC10, and SMAD13) with potential diagnostic and prognostic relevance. These genes, coupled with functional enrichment and interaction networks, deepen our understanding of GBM stem-like biology and pave the way for future translational research targeting glioblastoma stemness and therapy resistance.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Ethics Committee of Guilan University of Medical Sciences, Rasht, Iran (Code: IR.GUMS.REC.1403.037).
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Investigation, writing original draft of the manuscript, and bioinformatics analysis: Haniye Abassi and Saman Morovat; Investigation and methodology: Shahrokh Yousefzadeh-Chabok, Fatemeh Nejatifar, Kourosh Delpasand, and Seyedeh Elham Norollahi; Data collection: Afshin Dalili and Kosar Babaei; Reviewing, and editing: Ali Akbar Samadani, Shahrokh Yousefzadeh-Chabok, Fatemeh Nejatifar, Kourosh Delpasand, and Seyedeh Elham Norollahi; Conceptualization and supervision: Ali Akbar Samadani; Final approval: All authors.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgements
The authors would like to offer their special thanks to all people who were involved in this project.
References
Glioblastoma multiforme (GBM) is the most aggressive and lethal primary brain tumor in adults, accounting for nearly half of all malignant gliomas [1, 2]. Despite current multimodal treatments, including maximal surgical resection, radiotherapy, and temozolomide chemotherapy, median survival remains poor at approximately 12–15 months, and one-year survival rates are under 5–10% [3]. GBM can arise de novo (primary GBM) or evolve from lower-grade astrocytomas (secondary GBM), with primary cases comprising over 90% and usually affecting older individuals (~55–60 years) [4] without clinical or histologic evidence of a less malignant precursor lesion (primary glioblastomas.
Distinct genetic alterations, including IDH mutations, EGFR amplification, TERT promoter mutations, and MGMT promoter methylation, have been implicated in the pathogenesis and prognosis of GBM. In addition, intratumoral heterogeneity, immune evasion, and the restrictive blood–brain barrier further complicate treatment and contribute to the poor clinical outcome [5, 6]. As such, novel diagnostic markers and therapeutic strategies remain urgently needed.
A subpopulation of glioma stem-like cells (GSCs) (also referred to as cancer stem cells [CSCs]) has emerged as a key driver of GBM recurrence, resistance to therapy, and aggressive behavior. These cells display enhanced self-renewal and tumorigenic potential, and are enriched in hypoxic microenvironments characterized by necrosis and pseudopalisading architecture. Hypoxia-inducible factors, particularly HIF1α and HIF2α, promote CSC maintenance by activating downstream stem cell regulators such as SOX2, OCT4, and CD133, thereby supporting GBM progression [7, 8]. Moreover, other molecular processes (like EMT, epigenetic reprogramming, m6A RNA methylation, and expression of lncRNA H19) also contribute to GSC regulation and therapeutic resistance, underscoring the complexity of GBM biology [8–10].
Current evidence suggests that targeting pathways central to GSC function, such as hypoxia signaling, VEGF, TGF-β, CXCR4, and Notch, hold promise for improving therapeutic outcomes in GBM [6, 7]. However, the translation of CSC-targeted therapies remains challenging due to intratumoral heterogeneity and dynamic interactions within the tumor microenvironment.
Given this context, bioinformatics approaches that integrate gene expression profiling from GBM stem‑like cell models and clinical datasets may uncover novel biomarkers and therapeutic targets. In the current study, we analyze the GEO dataset GSE23806 (comprising glioblastoma stem‑like cell lines and conventional glioma lines) as well as TCGA GBM expression data via the GEPIA2 platform. By identifying common dysregulated genes, examining their prognostic significance, and mapping their protein‑interaction networks, we aim to discover novel diagnostic and survival‑related biomarkers associated with GBM stem‑like biology.
2. Methods and Materials/Patients
Data acquisition and preprocessing
The microarray dataset GSE23806 was downloaded from the Gene Expression Omnibus (GEO) [11] database using the GEOquery package [12, 13] in R software, version 4.2.2. This dataset, based on the Affymetrix Human Genome U133 Plus 2.0 Array platform, includes gene expression profiles from 32 conventional glioma cell lines and 23 glioblastoma stem-like cell lines. The glioblastoma stem-like group comprised 12 GS cell lines, 7 subclonal lines derived from two GS lines, 12 primary tumor samples, and 4 monolayer cultures established from tumors analogous to the GS lines.
Initial quality control and normalization of raw expression data were performed to reduce technical variation and ensure sample comparability. Normalization was confirmed through standard quality assessment metrics and visualizations using boxplots and density plots in R.
Differential expression analysis
To identify differentially expressed genes (DEGs) between glioblastoma stem-like cell lines and conventional glioma cell lines, the limma package from Bioconductor was employed. Genes with a |log₂ fold change ≥1| and an adjusted P<0.01 (Benjamini-Hochberg method) were considered significant.
To validate the results from GSE23806, the Gene Expression Profiling Interactive Analysis 2 (GEPIA2) web server was used to compare gene expression between glioblastoma (GBM) tissues and normal brain tissues, utilizing data from the cancer genome atlas (TCGA). Differentially expressed genes with the same threshold criteria (log₂ fold change ≥1, adj. P<0.01) were retrieved. The intersection of dysregulated genes from both datasets was determined to identify consistently differentially expressed genes in glioblastoma.
Survival analysis
The Kaplan–Meier survival analysis was conducted using GEPIA2 to assess the prognostic significance of the intersected dysregulated genes (n=355). Overall survival (OS) analysis was performed by stratifying patients into high- and low-expression groups based on median gene expression levels. Genes with a log-rank P<0.05 and a Hazard ratio (HR)>0.5 were considered significantly associated with survival outcomes. A manual literature review was conducted using PubMed to identify the top survival-related genes and assess their novelty in the context of GBM.
Protein–protein interaction (PPI) network construction
To explore the functional interactions among survival-associated genes, a PPI network was constructed using the GeneMANIA [14] plugin within the Cytoscape software, version 3.9 [15]. GeneMANIA integrates various data types, including physical protein interactions, co-expression, genetic interactions, co-localization, shared pathways, and protein domain similarity. The network was visualized and analyzed to identify gene clusters and patterns of connectivity.
Functional enrichment analysis
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analyses were performed using the clusterProfiler package [16] in R. The groupGO, enrichGO, and enrichKEGG functions were used to identify significantly enriched biological processes (BPs), molecular functions (MFs), cellular components (CCs), and pathways among the survival-related genes. A q<0.01 (adjusted using the Benjamini-Hochberg method) was set as the threshold for statistical significance.
3. Results
Dataset description and preprocessing
To identify DEGs, the microarray dataset GSE23806 was obtained from the GEO database using the GEOquery package in R. This dataset is based on the Affymetrix human genome U133 Plus 2.0 Array and includes expression profiles from 55 samples, comprising 32 conventional glioma cell lines, and 23 glioblastoma stem-like cell lines (12 GS cell lines, 7 subclonal lines derived from two GS lines, 12 primary tumors, and 4 monolayer cultures established from tumors similar to the GS lines). Before downstream analysis, the dataset underwent quality assessment and normalization to ensure comparability and minimize technical variation (Figure 1).
Identification of dysregulated genes in stem-like and tumor samples
Differential gene expression analysis was conducted using the limma package in R, with thresholds set at a log₂ fold change of ≥1 and an adjusted P<0.01. Based on this, 1211 dysregulated genes were identified in glioblastoma stem-like cell lines compared to conventional glioma cell lines.
To validate and extend these findings, the GEPIA2 web server was utilized to analyze gene expression in TCGA GBM samples in comparison to normal brain tissues. This analysis revealed 5217 genes that were dysregulated in tumor samples. The intersection of the two datasets yielded 355 commonly dysregulated genes, indicating a robust set of glioblastoma-associated transcripts (Figure 2).
Out of 1211 genes dysregulated in GSE23806 and 5217 in GEPIA2, 355 genes were commonly dysregulated, indicating strong concordance between stem-like cell line and tumor tissue expression profiles.
Survival analysis and identification of novel prognostic genes
A Kaplan–Meier survival analysis was performed using GEPIA2 to evaluate the association between OS and the 355 commonly dysregulated genes. As a result, 12 genes were found to have a statistically significant impact on patient survival.
A literature search was subsequently performed via PubMed to assess prior investigations of these 12 genes in glioblastoma. Six of these genes were identified as novel, with no prior evidence linking them to GBM Figure 3, Table 1).
These genes exhibit downregulated expression in GBM tissues compared to stem cell tissues, suggesting their potential as novel diagnostic and prognostic biomarkers for glioblastoma.
Construction of PPI network
To explore the molecular relationships among the 12 survival-related genes, a PPI network was constructed using the GeneMANIA plugin within cytoscape. GeneMANIA incorporates multiple functional datasets, including physical interactions, co-expression, pathways, and shared protein domains.
The resulting network comprised 26 nodes and 256 edges, representing extensive functional connectivity among the survival-associated genes (Figure 4).
The PPI network reveals a high degree of interconnectivity among the selected genes, indicating potential co-regulatory or synergistic roles in glioblastoma pathogenesis.
Functional enrichment analysis
To gain insight into the biological roles of the survival-related genes, functional enrichment analysis was performed using the clusterProfiler package in R. GO terms and KEGG pathways were assessed using groupGO, enrichGO, and enrichKEGG, with q<0.01 set as the threshold for significance.
This analysis revealed significant enrichment in pathways and BPs related to glioblastoma progression (Figure 5).
Key enriched GO terms and KEGG pathways highlight the involvement of these genes in critical BPs and signaling pathways relevant to glioblastoma stem cell maintenance and tumorigenesis.
4. Discussion
In this study, we identified 355 genes that are consistently dysregulated in both glioblastoma stem-like cell lines (GSE23806) and TCGA GBM tumor tissues, as determined by GEPIA2, of which 12 genes were significantly associated with overall survival. Remarkably, six genes (APLP1, CA14, PTPRN2, POR, ARMC10, and SMAD13) emerged as novel prognostic markers in GBM, to our knowledge, not previously reported in the context of glioblastoma literature.
In this study, we identified six genes (APLP1, CA14, PTPRN2, POR, ARMC10, and SMAD13) that are dysregulated in glioblastoma compared to stem cells and are significantly associated with patient survival. Based on HRs, two genes (CA14 and SMAD13) appear protective (HR<1), while the other four (APLP1, PTPRN2, POR, and ARMC10) are associated with increased risk (HR>1).
High-risk genes with HR>1 may contribute to more aggressive tumor behavior or stem-like characteristics, driving poorer outcomes. In contrast, protective genes with HR<1 (CA14 and SMAD13) suggest potential tumor-suppressive roles or involvement in pathways that moderate malignancy under hypoxic or stress conditions. This dichotomy reveals the biological complexity of GBM and might highlight context-dependent gene functions, such as differential roles in tumor initiation, progression, or response to therapy.
Although APLP1 has been implicated in neural development and amyloid processing, its diagnostic and prognostic value in GBM is largely unexplored. A recent study found that APLP1-positive extracellular vesicles (EVs) in patient plasma were significantly elevated in GBM, suggesting potential utility as a non-invasive biomarker for early detection [17]. Additionally, network-based analyses have highlighted APLP1, among other genes, as novel candidate biomarkers in GBM pathogenesis. These reports lend credence to our identification of APLP1 as a survival-related gene, supporting further investigation of its role in GBM biology.
CA14 encodes carbonic anhydrase XIV, which plays a crucial role in pH regulatory function, which is essential1818 for maintaining maintaining tumor microenvironment homeostasis under hypoxia and acidosis. Interestingly, CA14 has been identified by machine learning models as a key circadian rhythm gene predictive of melanoma prognosis, providing precedent for its potential as a diagnostic marker across malignancies. Its HR of 0.53 in our GBM cohort suggests that elevated CA14 may contribute to a more regulated microenvironment and improved survival, meriting further investigation in brain tumors [18-20].
POR (cytochrome P450 oxidoreductase) supports metabolic functions essential for the survival of tumor cells. Its association with poorer prognosis (HR=1.7) aligns with the metabolic reprogramming characteristic of aggressive GBM.
ARMC10 knockdown significantly reduced proliferation, migration, invasion, and tumor growth in GBM models. Gene set enrichment associates ARMC10 with Notch signaling and fatty acid metabolism, and the lipid reductions observed upon knockdown underscore its role in metabolic adaptation. Low ARMC10 levels correlated with better patient outcomes, reinforcing its potential as an adverse prognostic marker and therapeutic target.
SMAD13 (like other SMAD family proteins) modulates TGF-β signaling, a pathway heavily implicated in GBM aggression and the mesenchymal subtype. Although less studied than SMAD4 or Smad3, SMAD13 may similarly influence tumor growth, apoptosis, and differentiation. Its HR of 0.56 suggests a possible tumor-suppressive or differentiative role in GBM, potentially antagonizing mesenchymal transition [21-23].
Our PPI network, constructed using GeneMANIA, revealed 26 nodes and 256 edges, underscoring the extensive functional interplay among survival-related genes. Here, the network highlights functional associations, such as pathway co-expression and shared protein domains, suggesting that the novel genes are embedded within broader modules relevant to GBM. Future hub-centric analyses or cluster detection may pinpoint key regulatory modules that are amenable to therapeutic targeting.
The identification of six novel survival‑associated genes provides new avenues for diagnostic and therapeutic research. For instance, APLP1 may be measurable in EVs from blood, representing a minimally invasive diagnostic or monitoring tool. Other genes could serve as therapeutic targets, especially if they mediate stem-like or survival pathways in GBM. However, our findings are based on bioinformatics analyses, and further experimental validation in patient cohorts and functional assays is required. Future studies should evaluate whether these genes are expressed in glioma stem cell populations, respond to hypoxia or treatment, and affect tumor progression in xenograft or spheroid models.
A critical limitation of our work is the absence of experimental validation. Our findings are entirely based on bioinformatics and database-derived analyses and have not yet been corroborated through laboratory assays, such as RT-qPCR, Western blotting, immunohistochemistry, or functional characterization in vitro or in vivo.
This limitation mirrors a common challenge in bioinformatics-based biomarker discovery. For instance, prior studies employing integrated expression and survival analyses often emphasized the need for subsequent laboratory validation to confirm predicted biomarkers [24, 25]. Similarly, translational model investigations have shown that while in silico models may yield promising targets, only through direct experimental validation (such as gene knockdown or protein-level validation) can translational relevance be established [26].
5. Conclusion
By integrating stem-like GBM cell line data with tumor expression profiles and survival analysis, this study uncovered novel biomarker candidates (notably APLP1, CA14, PTPRN2, POR, ARMC10, and SMAD13) with potential diagnostic and prognostic relevance. These genes, coupled with functional enrichment and interaction networks, deepen our understanding of GBM stem-like biology and pave the way for future translational research targeting glioblastoma stemness and therapy resistance.
Ethical Considerations
Compliance with ethical guidelines
This study was approved by the Ethics Committee of Guilan University of Medical Sciences, Rasht, Iran (Code: IR.GUMS.REC.1403.037).
Funding
This research did not receive any grant from funding agencies in the public, commercial, or non-profit sectors.
Authors' contributions
Investigation, writing original draft of the manuscript, and bioinformatics analysis: Haniye Abassi and Saman Morovat; Investigation and methodology: Shahrokh Yousefzadeh-Chabok, Fatemeh Nejatifar, Kourosh Delpasand, and Seyedeh Elham Norollahi; Data collection: Afshin Dalili and Kosar Babaei; Reviewing, and editing: Ali Akbar Samadani, Shahrokh Yousefzadeh-Chabok, Fatemeh Nejatifar, Kourosh Delpasand, and Seyedeh Elham Norollahi; Conceptualization and supervision: Ali Akbar Samadani; Final approval: All authors.
Conflict of interest
The authors declared no conflict of interest.
Acknowledgements
The authors would like to offer their special thanks to all people who were involved in this project.
References
- Zheng X, Tang Q, Ren L, Liu J, Li W, Fu W, et al. A narrative review of research progress on drug therapies for glioblastoma multiforme. Annals of Translational Medicine. 2021; 9(11):943.[DOI:10.21037/atm-20-8017] [PMID]
- Friedman HS, Bigner DD. Glioblastoma multiforme and the epidermal growth factor receptor. The New England Journal of Medicine. 2005; 353(19):1997-9. [DOI:10.1056/NEJMp058186] [PMID]
- Sipos D, Raposa BL, Freihat O, Simon M, Mekis N, Cornacchione P, et al. Glioblastoma: Clinical presentation, multidisciplinary management, and long-term outcomes. Cancers. 2025; 17(1):146. [DOI:10.3390/cancers17010146] [PMID]
- Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clinical Cancer Research. 2013; 19(4):764-72. [DOI:10.1158/1078-0432.CCR-12-3002] [PMID]
- Rong L, Li N, Zhang Z. Emerging therapies for glioblastoma: current state and future directions. Journal of Experimental & Clinical Cancer Research. 2022; 41(1):142. [DOI:10.1186/s13046-022-02349-7] [PMID]
- Roos A, Ding Z, Loftus JC, Tran NL. Molecular and microenvironmental determinants of glioma stem-like cell survival and invasion. Frontiers in Oncology. 2017; 7:120. [DOI:10.3389/fonc.2017.00120] [PMID]
- Boyd NH, Tran AN, Bernstock JD, Etminan T, Jones AB, Gillespie GY, et al. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics. 2021; 11(2):665-83. [DOI:10.7150/thno.41692] [PMID]
- Abd GM, Laird MC, Ku JC, Li Y. Hypoxia-induced cancer cell reprogramming: A review on how cancer stem cells arise. Frontiers in Oncology. 2023; 13:1227884. [DOI:10.3389/fonc.2023.1227884] [PMID]
- Morovat S, Morovat P, Kalantary-Charvadeh A, Mojbafan M, Teimourian S. LINC01410 in cancer: A comprehensive review of its oncogenic role, regulatory mechanisms, and clinical implications. Medical Oncology. 2025; 42(8):325. [DOI:10.1007/s12032-025-02889-w] [PMID]
- Kalantary-Charvadeh A, Morovat S, Aslani S, Ziamajidi N, Emami Razavi A, Abbasalipourkabir R. The role of long non-coding RNA LINC00839 in oral squamous cell carcinoma based on bioinformatics and experimental research. Scientific Reports. 2024 ;14(1):31817. [DOI:10.1038/s41598-024-82922-6] [PMID]
- Clough E, Barrett T. The gene expression omnibus database. Methods in Molecular Biology. 2016; 1418:93-110. [DOI:10.1007/978-1-4939-3578-9_5] [PMID]
- Davis S, Meltzer PS. GEOquery: A bridge between the gene expression omnibus (GEO) and bioconductor. Bioinformatics. 2007; 23(14):1846-7. [DOI:10.1093/bioinformatics/btm254] [PMID]
- Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Research. 2019; 47(W1):W556-60. [DOI:10.1093/nar/gkz430] [PMID]
- Franz M, Rodriguez H, Lopes C, Zuberi K, Montojo J, Bader GD, et al. GeneMANIA update 2018. Nucleic Acids Research. 2018; 46(W1):W60-4. [DOI:10.1093/nar/gky311] [PMID]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research. 2003; 13(11):2498-504. [DOI:10.1101/gr.1239303] [PMID]
- Yu G, Wang LG, Han Y, He QY. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 2012; 16(5):284-7. [DOI:10.1089/omi.2011.0118] [PMID]
- Choi Y, Park JH, Jo A, Lim CW, Park JM, Hwang JW, et al. Blood-derived APLP1+ extracellular vesicles are potential biomarkers for the early diagnosis of brain diseases. Science Advances. 2025; 11(1):eado6894. [DOI:10.1126/sciadv.ado6894] [PMID]
- Liu M, Yu Y, Zhang Z, Chen Z, Chen B, Cheng Y, et al. AEBP1 as a potential immune-related prognostic biomarker in glioblastoma: A bioinformatic analyses. Annals of Translational Medicine. 2021; 9(22):1657. [DOI:10.21037/atm-21-5183] [PMID]
- Wang C, Han H, Cheng F, et al. Clinical significance and potential mechanism of AEBP1 in glioblastoma. Journal of Neuropathology and Experimental Neurology. 2024; 83(12):1020-9. [DOI:10.1093/jnen/nlae091] [PMID]
- Alshabi AM, Vastrad B, Shaikh IA, Vastrad C. Identification of crucial candidate genes and pathways in glioblastoma multiform by bioinformatics analysis. Biomolecules. 2019; 9(5):201. [DOI:10.3390/biom9050201] [PMID]
- Kim Y, Varn FS, Park SH, Yoon BW, Park HR, Lee C, et al. Perspective of mesenchymal transformation in glioblastoma. Acta Neuropathologica Communications. 2021; 9(1):50. [DOI:10.1186/s40478-021-01151-4] [PMID]
- Feng B, Gao T, Chen L, Xing Y. ARMC10 drives glioblastoma progression through activating notch pathway. Molecular Carcinogenesis. 2025; 64(5):883-96. [DOI:10.1002/mc.23895] [PMID]
- Zhao M, Mishra L, Deng CX. The role of TGF-β/SMAD4 signaling in cancer. International Journal of Biological Sciences. 2018; 14(2):111-23. [DOI:10.7150/ijbs.23230] [PMID]
- Ali H, Harting R, de Vries R, Ali M, Wurdinger T, Best MG. Blood-based biomarkers for glioma in the context of gliomagenesis: A systematic review. rontiers in Oncology. 2021; 11:665235. [DOI:10.3389/fonc.2021.665235] [PMID]
- Huang Z, Chen Z, Song E, Yu P, Chen W, Lin H. Bioinformatics analysis and experimental validation for exploring key molecular markers for glioblastoma. Applied Biochemistry and Biotechnology. 2024; 196(10):6974-92. [DOI:10.1007/s12010-024-04894-7] [PMID]
- Ahmadi-Beni R, Shahbazi S, Khoshnevisan A. An integrative bioinformatics investigation and experimental validation of critically involved genes in high-grade gliomas. Diagnostic Pathology. 2022; 17(1):73. [DOI:10.1186/s13000-022-01253-0] [PMID]
Type of Study: Research |
Subject:
Brain Tumors
Send email to the article author
| Rights and Permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright © The Author(s);
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-By-NC), which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Contact Information
